Testosterone and Prostate Cancer - Bye Androgen Hypothesis, Welcome Saturation Model

Key Points
What is known - The Androgen Hypothesis
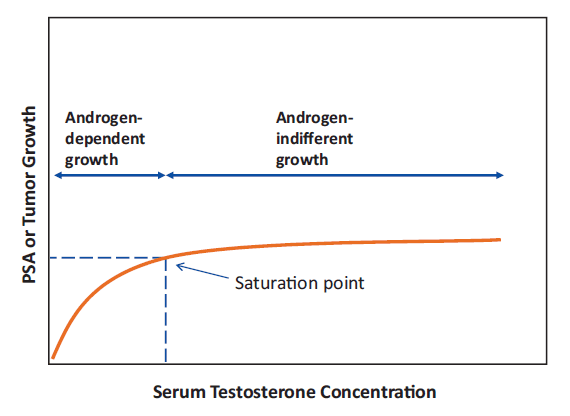
What this study adds - The Saturation Model
Summary
1. Gooren, L.J. and H.M. Behre, Diagnosing and treating testosterone deficiency in different parts of the world: changes between 2006 and 2010. Aging Male, 2012. 15(1): p. 22-7.
2. Atan, A., et al., Serum testosterone level, testosterone replacement treatment, and prostate cancer. Adv Urol, 2013. 2013: p. 275945.
3. Khera, M., et al., A new era of testosterone and prostate cancer: from physiology to clinical implications. Eur Urol, 2014. 65(1): p. 115-23.
4. Huggins, C. and C.V. Hodges, The effect of castration, of estrogen and of androgen injection on serum phosphatase in metastatic carcinoma of the prostate. Cancer Res 1941;1:293–7, 1941.
5. Huggins, C., R.E.J. Stevens, and C.V. Hodges, The effect of castration, of estrogen and of androgen injection on serum phosphatase in metastatic carcinoma of the prostate. Arch Surg 1941;43:209-223, 1941.
6. Morgentaler, A., Guilt by association: a historical perspective on Huggins, testosterone therapy, and prostate cancer. J Sex Med, 2008. 5(8): p. 1834-40.
7. Roddam, A.W., et al., Endogenous sex hormones and prostate cancer: a collaborative analysis of 18 prospective studies. J Natl Cancer Inst, 2008. 100(3): p. 170-83.
8. Calof, O.M., et al., Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci, 2005. 60(11): p. 1451-7.
9. Fernandez-Balsells, M.M., et al., Clinical review 1: Adverse effects of testosterone therapy in adult men: a systematic review and meta-analysis. J Clin Endocrinol Metab, 2010. 95(6): p. 2560-75.
10. Feneley, M.R. and M. Carruthers, Is testosterone treatment good for the prostate? Study of safety during long-term treatment. J Sex Med, 2012. 9(8): p. 2138-49.
11. Raynaud, J.P., et al., Prostate-specific antigen (PSA) concentrations in hypogonadal men during 6 years of transdermal testosterone treatment. BJU Int, 2013. 111(6): p. 880-90.
12. Wang, C., et al., Investigation, treatment, and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA, and ASA recommendations. Eur Urol, 2009. 55(1): p. 121-30.
13. Morgentaler, A., Rapidly shifting concepts regarding androgens and prostate cancer. ScientificWorldJournal, 2009. 9: p. 685-90.
14. Garcia-Cruz, E., et al., Low testosterone level predicts prostate cancer in re-biopsy in patients with high grade prostatic intraepithelial neoplasia. BJU Int, 2012. 110(6 Pt B): p. E199-202.
15. Garcia-Cruz, E., et al., Low testosterone levels are related to poor prognosis factors in men with prostate cancer prior to treatment. BJU Int, 2012. 110(11 Pt B): p. E541-6.
16. Brawn, P.N. and V.O. Speights, The dedifferentiation of metastatic prostate carcinoma. Br J Cancer, 1989. 59(1): p. 85-8.
17. Hatzoglou, A., et al., Membrane androgen receptor activation induces apoptotic regression of human prostate cancer cells in vitro and in vivo. J Clin Endocrinol Metab, 2005. 90(2): p. 893-903.
18. Sonnenschein, C., et al., Negative controls of cell proliferation: human prostate cancer cells and androgens. Cancer Res, 1989. 49(13): p. 3474-81.
19. Chuu, C.P., et al., Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res, 2005. 65(6): p. 2082-4.
20. Kava, B.R., To treat or not to treat with testosterone replacement therapy: a contemporary review of management of late-onset hypogonadism and critical issues related to prostate cancer. Curr Urol Rep, 2014. 15(7): p. 422.
21. Schroder, F., et al., Androgen deprivation therapy: past, present and future. BJU Int, 2012. 109 Suppl 6: p. 1-12.
22. Monath, J.R., et al., Physiologic variations of serum testosterone within the normal range do not affect serum prostate-specific antigen. Urology, 1995. 46(1): p. 58-61.
23. Monda, J.M., et al., The correlation between serum prostate-specific antigen and prostate cancer is not influenced by the serum testosterone concentration. Urology, 1995. 46(1): p. 62-4.
24. Bhasin, S., et al., The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med, 1996. 335(1): p. 1-7.
25. Cooper, C.S., et al., Effect of exogenous testosterone on prostate volume, serum and semen prostate specific antigen levels in healthy young men. J Urol, 1998. 159(2): p. 441-3.
26. Behre, H.M., J. Bohmeyer, and E. Nieschlag, Prostate volume in testosterone-treated and untreated hypogonadal men in comparison to age-matched normal controls. Clin Endocrinol (Oxf), 1994. 40(3): p. 341-9.
27. Meikle, A.W., et al., Prostate size in hypogonadal men treated with a nonscrotal permeation-enhanced testosterone transdermal system. Urology, 1997. 49(2): p. 191-6.
28. Moon, D.G., et al., The efficacy and safety of testosterone undecanoate (Nebido((R))) in testosterone deficiency syndrome in Korean: a multicenter prospective study. J Sex Med, 2010. 7(6): p. 2253-60.
29. Snyder, P.J., et al., Effects of testosterone replacement in hypogonadal men. J Clin Endocrinol Metab, 2000. 85(8): p. 2670-7.
30. Wang, C., et al., Long-term testosterone gel (AndroGel) treatment maintains beneficial effects on sexual function and mood, lean and fat mass, and bone mineral density in hypogonadal men. J Clin Endocrinol Metab, 2004. 89(5): p. 2085-98.
31. Morgentaler, A. and A.M. Traish, Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth. Eur Urol, 2009. 55(2): p. 310-20.
32. Marks, L.S., et al., Effect of testosterone replacement therapy on prostate tissue in men with late-onset hypogonadism: a randomized controlled trial. JAMA, 2006. 296(19): p. 2351-61.
33. Morgentaler, A., et al., Testosterone therapy in men with untreated prostate cancer. J Urol, 2011. 185(4): p. 1256-60.

